
Summary
- Data integrity is the foundation of every regulatory submission, batch release decision, and patient safety determination — defined by the FDA as the “completeness, consistency, and accuracy of data” across its entire lifecycle.
- ALCOA++ framework (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available, and Traceable) is the de facto universal standard, explicitly referenced across FDA, EU Annex 11, PIC/S, and WHO guidelines.
- FDA 21 CFR Part 11 and EU GMP Annex 11 together establish the rules for electronic records, unique user authentication, system-generated audit trails, validated backups, and controls preventing unauthorized record alteration.
- Most data integrity failures are structural, not behavioral — shared login credentials, deleted raw data, unvalidated hybrid systems, and inadequate audit trail review account for the majority of FDA Warning Letter citations.
- Digital validation platforms like Kneat Gx eliminate root-cause data integrity risks by design — delivering 60% reductions in validation cycle times, 40% reductions in protocol review and approval time, and full alignment with 21 CFR Part 11, EU Annex 11, and ALCOA++.
Data integrity in life sciences is the regulatory and operational foundation that determines whether product safety decisions can be trusted. Failures have resulted in FDA Warning Letters, import alerts, and product recalls. The ALCOA++ framework is the universal global standard embedded across FDA, EU Annex 11, and PIC/S requirements. It covers ten principles: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available, and Traceable.The most common failure points are structural — shared credentials, deleted raw data, and unvalidated systems — not isolated human errors. Purpose-built digital validation platforms eliminate these root-cause risks by design.
Data integrity failures carry real consequences. A single falsified batch record, a deleted chromatography file, or a shared login credential can trigger an FDA Warning Letter, halt a product release, or put patients at risk. This guide to data integrity in life sciences exists because those consequences are preventable — and because the quality, validation, and compliance professionals responsible for preventing them deserve a clear, practitioner-focused resource.
Kneat was founded by pharmaceutical engineers who understood these challenges firsthand. Today, we work with eight of the world’s top 10 biopharma companies. This resource is built for professionals operating in GxP environments across pharmaceutical, biotechnology, and medical device organizations. Whether you manage validation programs, oversee quality systems, or lead compliance strategy, this guide covers the regulatory landscape, the ALCOA++ framework, common failure points, and the practical role digital validation plays in building a program that holds up under scrutiny.
What is data integrity in life sciences and why does it matter?
Data integrity in life sciences is the foundation upon which every regulatory submission, batch release decision, and patient safety determination rests. The FDA defines it precisely: data integrity means “the completeness, consistency, and accuracy of data.” That definition, drawn from the FDA’s 2018 CGMP Data Integrity Guidance, sets the compliance baseline for every pharmaceutical, biotech, and medical device organization operating in a regulated environment.
The Pharmaceutical Inspection Co-operation Scheme (PIC/S) extends this further, defining data integrity as “the degree to which data are complete, consistent, accurate, trustworthy, and reliable throughout the data lifecycle.” The scope is broad and intentional. Data integrity applies to all records — paper and electronic — generated across R&D, manufacturing, clinical trials, and regulatory submissions.
GxP data integrity failures carry direct, documented consequences. Over the past decade, FDA Warning Letters citing data integrity violations have increased substantially, spanning issues from deleted raw data to backdated laboratory records. Those violations trigger import alerts, product recalls, and consent decrees. Reputational damage compounds the operational cost.
The regulatory logic is straightforward: if the data supporting a product’s safety and efficacy cannot be trusted, neither can the product. A single compromised record in a batch manufacturing file creates downstream risk that extends to patients. This is not a theoretical concern — regulators have demonstrated consistent willingness to act, and the consequences of non-compliance far outweigh the investment required to build a robust data integrity program.

The regulatory framework — What the FDA and EU require
Understanding what data integrity demands in practice starts with understanding where those demands originate. FDA 21 CFR Part 11 compliance sits at the center of any global data integrity program, but it is one of three interlocking regulatory frameworks that life sciences manufacturers must satisfy simultaneously.
21 CFR Part 11 governs electronic records and electronic signatures in FDA-regulated environments. It requires unique user authentication, system-generated audit trails, and controls preventing unauthorized record alteration. Paired with 21 CFR §211.68(b), which mandates that computerized systems produce accurate and complete records, these regulations establish the baseline for electronic data integrity in US manufacturing operations.
EU GMP Annex 11 addresses computerized systems across the full data lifecycle. Clause 9 requires that data entered into a computerized system must be checked for accuracy. It also requires that audit trails capture all Current Good Manufacturing Practice (CGMP)-relevant changes, including the original entry, the reason for change, and the identity of the person making it. Annex 11 also mandates validated backup and recovery procedures to ensure data availability.
A critical point: the FDA’s 2018 CGMP Data Integrity Guidance explicitly places responsibility on senior management, not IT teams alone. Culture, oversight, and organizational accountability are regulatory requirements, not optional governance practices.
Key Requirements Across the FDA and EU
The table below maps each major regulatory framework to its core data integrity requirements. Use this as a quick reference when assessing compliance obligations across jurisdictions.
| Requirement | FDA (21 CFR Part 11 / Parts 210–211) | EU GMP Annex 11 |
|---|---|---|
| Access controls | Unique user IDs and passwords required; system access limited by role (21 CFR §11.10(d)) | Authorized access only; physical and logical controls required (Annex 11, Clause 12) |
| Audit trails | Computer-generated, time-stamped audit trails capturing record creation, modification, and deletion (21 CFR §11.10(e)) | Audit trails must record all GMP-relevant changes; reason for change required (Annex 11, Clause 9) |
| Electronic signatures | Legally binding; must link signature to record; include printed name, date, and meaning (21 CFR §11.50) | Electronic signatures must be equivalent to handwritten signatures; controlled and verified (Annex 11, Clause 14) |
| Data backup and recovery | Accurate and complete copies of records must be maintained and retrievable (21 CFR §211.68(b)) | Regular backups required; backup integrity and restoration must be tested (Annex 11, Clause 7.1) |
| System validation | Computerized systems must be validated to demonstrate accuracy, reliability, and consistent performance (21 CFR §11.10(a)) | Systems must be validated before use and revalidated after significant changes (Annex 11, Clause 4) |
Global manufacturers operating across multiple jurisdictions must satisfy all three frameworks simultaneously. Requirements overlap significantly, but specific language and emphasis differ. Mapping your systems to all three frameworks from the outset reduces remediation risk and simplifies regulatory inspection readiness.
The ALCOA++ Framework — The Universal standard for data integrity
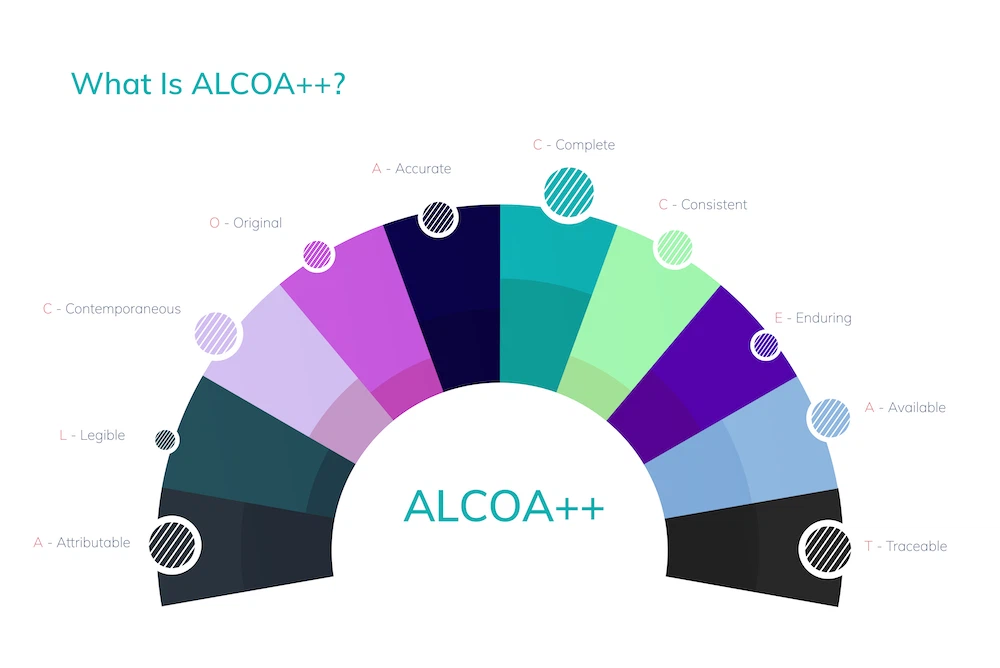
ALCOA++ is the de facto universal global standard for data integrity. It is explicitly referenced in PIC/S PI 041-1, FDA CGMP Guidance, EU Annex 11, and WHO GMP guidelines across all major regulatory jurisdictions. The original five ALCOA principles were introduced in the 1990s. Industry practice extended the framework with four additional attributes by 2010, addressing the complexity of electronic systems and global supply chains. Traceability was added more recently, completing ALCOA++.
Here is what each attribute means in practice:
- Attributable: Every data entry must identify who recorded it and when. Shared login credentials violate this principle directly — and remain one of the most cited data integrity failures in FDA Warning Letters.
- Legible: Records must be readable for their entire retention period. Faded handwritten entries on a batch record or corrupted file formats both fail this requirement.
- Contemporaneous: Data must be recorded at the time the activity occurs. Pre-recording or back-dating test results — even under schedule pressure — constitutes a direct violation.
- Original: The first-captured record is the authoritative record. Discarding raw chromatography data and retaining only transcribed summaries destroys the original and eliminates the ability to verify results.
- Accurate: Data must reflect what actually occurred, without correction fluid, unauthorized deletions, or selective reporting of out-of-specification results.
- Complete: All data — including failed runs, voided entries, and anomalous results — must be retained. Incomplete records distort the full picture of a process or product.
- Consistent: Data entries must follow a logical, chronological sequence. Timestamps that contradict each other or entries recorded out of sequence raise immediate regulatory concern.
- Enduring: Records must remain intact and retrievable for their full required retention period. Storing critical data on a single unbackup server fails this requirement by design.
- Available: Authorized personnel and regulators must be able to access records promptly. Data archived in inaccessible legacy systems creates practical and regulatory risk simultaneously.
- Traceable: Data must have a complete, unbroken audit trail through its entire lifecycle.
How ALCOA++ applies to electronic records and audit trails
Electronic systems introduce specific audit trail requirements that pharma organizations must address at the system design level. Unique user authentication satisfies Attributable. System-generated, tamper-evident audit trails capture Complete and Consistent requirements automatically.
Dynamic record formats — not static PDF exports — preserve the Original record as required by FDA CGMP guidance, which explicitly states that printouts do not substitute for retaining original electronic records. Synchronized, server-controlled timestamps enforce Contemporaneous recording. Centralized, validated repositories with defined backup schedules satisfy both Enduring and Available.
These are not configuration options — they are baseline compliance requirements for any validated computerized system operating in a GxP environment.
Common data integrity challenges and what causes most failures
The most persistent GxP data integrity failures fall into three categories: system design flaws, process gaps, and organizational culture pressures. Most failures are not deliberate fraud — they are structural vulnerabilities that create conditions where data can be altered, lost, or misrepresented. In our experience working with global pharmaceutical manufacturers — including eight of the world’s top 10 biopharma companies — this pattern holds consistently across organization size and geography.
FDA Warning Letters consistently cite the same recurring violations. Shared login credentials remain among the most frequently observed failures. When multiple analysts use a single account, the Attributable requirement collapses entirely — the system cannot establish who performed an action, when, or under what circumstances. This is a design failure, not a behavioral one.
Deletion or overwriting of raw data without audit trail capture represents another critical risk. Instruments that allow users to rerun analyses and save over original results — without capturing the prior state — violate Original and Complete requirements under ALCOA++. FDA inspectors are specifically trained to detect anomalies in electronic system logs that indicate this pattern.
Paper-based and hybrid systems introduce a third category of vulnerability. Manual transcription between paper records and electronic systems creates gaps where data can be altered between capture and entry. Neither system holds a complete, verified record.
Case in point: MSD (Merck Sharp & Dohme) evaluated its own hybrid validation environment — a mix of paper protocols, Microsoft Word templates, SharePoint-based tools, and wet-ink signatures.
The advantage of Kneat is it gives us that end-to-end without paper. Things are contemporaneous, the reviews and approvals are concurrent. For us, it gives us that data integrity advantage that we didn’t have before, when some documents were in paper and some were electronic
Director of IT Compliance, Global Systems
MSD’s hybrid model is representative of the structural gap that makes data integrity violations structurally predictable, not exceptional.
Inadequate system validation compounds these risks. Electronic systems that have not been properly validated under FDA 21 CFR Part 11 compliance requirements may lack enforced access controls, leaving records manipulable after the fact.
Finally, production pressure can incentivize result manipulation — not always through deliberate fraud, but through normalized shortcuts: backdating entries, selecting favorable test results, or delaying documentation. These behaviors emerge from governance failures, not individual character flaws.
Building a data governance system that sustains compliance
A sustainable data governance program requires people, processes, and technology working simultaneously — not sequentially. PIC/S PI 041-1 defines data governance as “the sum total of arrangements which provide assurance of data integrity.”‘ Both PIC/S and FDA place senior management at the center of that system. Oversight and organizational culture are not separable from technical controls: a well-configured audit trail means little inside an organization where pressure to meet timelines discourages accurate reporting.
Data governance in life sciences is not a project with a completion date. A functional program requires five practical elements working together:
- Documented data lifecycle mapping — identify every point where data is generated, transferred, processed, or archived.
- Role-based access control and segregation of duties — ensure no single user can create, approve, and release their own records.
- Validated computerized systems with enabled audit trails — system validation confirms that CGMP workflows perform their intended function.
- Periodic self-inspection and audit trail review — build scheduled review into routine quality operations, not just pre-inspection preparation.
- Staff training on ALCOA++ and reporting expectations — personnel must understand both the principles and the consequences of non-compliance.
These elements reinforce one another. Mapping the data lifecycle reveals where access controls are weakest. Training without validated systems leaves gaps that no procedure can fully close.
Access controls, audit trails, and system validation
Three technical controls form the non-negotiable foundation of data governance in any GxP-regulated environment.
First, every user must authenticate individually. Shared credentials directly violate Attributable requirements and make it impossible to reconstruct who performed a specific action — individual logins are a baseline regulatory expectation, not an IT preference.
Second, systems must generate tamper-evident audit trails automatically, capturing who made a change, what changed, when it occurred, and the reason for the change. Meeting audit trail requirements in pharma means these records exist at the system level — not as manually maintained logs.
Third, computerized systems must be validated to confirm that CGMP workflows perform their intended functions reliably and consistently.
Critically, the FDA’s CGMP guidance specifies that audit trail review must be part of routine data review — conducted by quality personnel before batch release decisions, not delegated to periodic IT maintenance cycles. Organizations that treat audit trail review as an IT function rather than a quality function create a significant compliance gap.
How digital validation platforms eliminate root-cause data integrity risks
Paper-based and hybrid validation systems do not fail because people make mistakes — they fail because their architecture makes data integrity violations structurally inevitable. Handwritten records can be backdated. Spreadsheets can be edited without trace. Scanned PDFs preserve an image, not a dynamic record. These are not user behavior problems; they are design problems that governance frameworks and technical controls alone cannot fully resolve.
Purpose-built digital validation platforms eliminate these vulnerabilities at the source by embedding ALCOA++ compliance directly into system architecture. The key mechanisms are distinct:
- Automated user authentication ensures every action is Attributable to a specific, verified individual.
- Electronic records that lock on completion preserve dynamic content rather than static snapshots, satisfying both Original and Legible requirements.
- Real-time data capture during test execution eliminates the possibility of after-the-fact reconstruction, meeting the Contemporaneous standard.
- System-generated audit trails capture every entry, change, and approval automatically, fulfilling Complete and Consistent requirements without relying on manual logging.
- Centralized, encrypted cloud storage with configurable retention periods addresses the Enduring and Available attributes across global sites and regulatory jurisdictions.
Kneat Gx is built to satisfy both 21 CFR Part 11 and EU Annex 11 by design, not by workaround. The results from global deployments demonstrate what this structural approach delivers in practice: organizations using Kneat Gx report a 60% reduction in validation cycle times and have eliminated 46% of process steps entirely.
Critically, 97% of customers rate Kneat’s support as “Very Good” or “Excellent,” reflecting the operational reality of sustained adoption, not just initial implementation. Kneat Gx is also recognized as a Leader in three of G2’s Fall 2025 Grid® Reports for Pharma and Biotech, achieving a G2 Satisfaction Score of 98 out of 100 — 20 points higher than the next closest competitor.
Real-world example: Recipharm Advanced Bio, one of the world’s leading Contract Development and Manufacturing Organizations (CDMOs), faced exactly this challenge at its biologics manufacturing facility in Watertown, Massachusetts. With the FDA recording over 180 data integrity–related observations across the industry, the organization needed a system that could deliver full alignment with 21 CFR Part 11, EU Annex 11, and ALCOA++ principles — without slowing down quality operations.
After implementing Kneat Gx across Computer System Validation (CSV), Commissioning, Qualification and Validation (CQV), Analytical Instrument Qualification (AIQ), eForms, and electronic logbooks, review cycles dropped from days to hours. The platform became a centralized hub for drafting, review, approval, and QC sample tracking.
By digitalizing electronic logbooks, I don’t have to spend time creating or archiving paper logbooks anymore — everything is in one place, What used to take multiple systems and manual steps, we now do directly in Kneat.
Kailash Rathi, Director of Quality Systems & Validation at Recipharm Advanced Bio
A digital validation platform does not simply digitize paper. It removes the conditions that make data integrity failures possible in the first place.
The scale at which this structural approach delivers results is also documented across the largest pharmaceutical organizations in the world. A Top 10 global pharmaceutical and healthcare company deployed Kneat Gx for CQV and AIQ across multiple global sites as part of a corporate-wide paperless manufacturing initiative.
The results from a direct paper-versus-Kneat comparison at the pilot facility were unambiguous: a 40% reduction in protocol review and approval time, a 40% reduction in total IOQ calendar hours, and complete elimination of execution binder preparation. “Did we get value by implementing Kneat? It’s a no brainer. We’re seeing gains in schedule, gains in work hours, positive feedback from the user community, and improvements in the core metrics — it is an easy discussion to have,” said the company’s Global Engineering, C&Q lead.
Frequently asked questions about data integrity in life sciences
Q: What is the difference between data integrity and data quality in pharma?
Data integrity refers to whether data is attributable, complete, consistent, and unaltered across its entire lifecycle. Data quality refers to whether data is fit for purpose: accurate, precise, and meaningful for decision-making. Integrity is a prerequisite for quality. If integrity has been compromised, no data quality assessment can be considered reliable.
Q: What are the ALCOA++ principles, and are they legally required?
ALCOA++ stands for Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available, and Traceable. While the term “ALCOA++” does not appear verbatim in every regulation, its principles are embedded in FDA CGMP regulations, EU GMP Annex 11, and PIC/S Guidance PI 041-1. For any GxP-regulated operation, compliance with ALCOA++ principles is effectively mandatory.
Q: How often should audit trails be reviewed in a pharmaceutical facility?
Per FDA CGMP guidance, audit trail review frequency must align with data criticality and activity type. Audit trails supporting batch release decisions must be reviewed before release. For other systems, organizations must document a risk-based review schedule rather than deferring review to periodic IT assessments.
Q: What triggers an FDA data integrity inspection or Warning Letter?
Common triggers include unexplained data deletions, backdated entries, shared login credentials, and laboratory results that appear implausibly consistent. Whistleblower reports also prompt inspections. FDA investigators are specifically trained to identify anomalies in audit trails and electronic system logs.
Q: Can a digital validation platform replace a paper-based system for CGMP compliance?
Yes, provided the system is validated in accordance with 21 CFR Part 11 or EU Annex 11. Electronic records validated under these frameworks are legally equivalent to paper records. Purpose-built digital validation platforms such as Kneat Gx typically deliver stronger data integrity assurance than paper by eliminating manual transcription errors and enabling automatic audit trail capture.
Q: What is the difference between paper-based and electronic validation for GxP compliance?
Paper-based validation relies on manual processes — handwritten records, wet-ink signatures, and physical binder management — that create structural opportunities for data to be altered, backdated, or lost without trace. Electronic validation platforms validated under 21 CFR Part 11 and EU Annex 11 replace these manual steps with automated audit trails, enforced user authentication, and dynamic record locking. Organizations that have transitioned from paper to Kneat Gx have reported a 40% reduction in protocol review and approval time and complete elimination of execution binder preparation.
Q: How does a digital validation platform support ALCOA++ compliance?
A purpose-built digital validation platform operationalizes each ALCOA++ attribute at the system architecture level. Enforced individual user authentication satisfies Attributable. Real-time data capture during test execution satisfies Contemporaneous. Dynamic record locking on completion satisfies Original. System-generated, tamper-evident audit trails satisfy Complete and Consistent. Centralized cloud storage with validated retention schedules satisfies Enduring and Available. These controls are built into Kneat Gx by design, not applied as workarounds after implementation.
The foundation your quality program depends on
Data integrity is not a compliance checkbox. It is the structural condition that determines whether every quality decision, safety assessment, and regulatory submission your organization makes can be trusted. This guide to data integrity in life sciences has covered the ground that matters most: the regulatory frameworks that define the standard, the ALCOA++ principles that operationalize it, the governance systems that sustain it, and the failure patterns that most commonly undermine it.
Sustainable data integrity requires the right framework, the right governance, and the right technology working in concert — no single element is sufficient on its own. Organizations that treat these three dimensions as integrated, rather than sequential, are the ones that maintain compliance under inspection pressure and build quality programs that scale.
As the digital validation platform trusted by 8 of the world’s top 10 biopharma companies, Kneat Gx is purpose-built to operationalize data integrity across every validation use case. If you are ready to move from risk to readiness, Book a demo to see how Kneat Gx can help.



