
By understanding the most common reasons for receiving Form 483s and warning letters, companies operating in the life sciences industry can take necessary steps to prevent non-compliance and avoid the associated consequences.
Form 483s and warnings are regulatory compliance-related documents that are issued by the U.S. Food and Drug Administration (FDA) during inspections of facilities that manufacture drugs, medical devices, and other FDA-regulated products. These documents are a way for the FDA to communicate their observations and concerns about non-compliance with regulations, policies, and procedures to the inspected facility.
The consequences of not addressing Form 483s and warnings can include a delay in approval of new products, suspension or revocation of licenses, legal action, and damage to reputation, among others. Companies must take appropriate steps to ensure compliance with FDA regulations to avoid these consequences.
Download our Digital Validation Handbook to find out everything you need to know about how to streamline validation, leave paper-based processes behind, mitigate noncompliance risks, and drive more value for your company.
This article will explore some of the top reasons for receiving Form 483s and warnings, including:
- Inadequate procedures
- Poor record-keeping
- Lack of training
- Contamination and cross-contamination
- Failure to follow written procedures
What Is a form 483?
A Form 483 is a document issued by FDA inspectors after an inspection to communicate to the inspected facility the observations made during the inspection, which indicate possible violations of the Federal Food, Drug, and Cosmetic Act (FD&C Act) and related regulations. The purpose of a Form 483 is to notify the facility management of these observations, provide the facility with an opportunity to make necessary corrections, and to serve as a basis for further regulatory action if deemed necessary.

What Is a warning letter?
A warning letter is a formal communication from the FDA to a facility that has received a Form 483, indicating the agency has determined the violations identified during the inspection are serious enough to warrant regulatory action. The warning letter usually outlines the specific violations identified and requests the facility to take prompt corrective action to address the issues.
Common reasons for form 483s and warnings
The FDA issues Form 483s and warning letters when they find violations of regulations during inspections. These violations can range from inadequate procedures, poor record-keeping, and lack of employee training to contamination and cross-contamination, and failure to follow written procedures, among others.
1. Inadequate procedures
Inadequate procedures refer to procedures that are not adequate or appropriate to ensure compliance with FDA regulations. This can include a lack of procedures, incomplete or outdated procedures, or procedures not being followed correctly. Examples of inadequate procedures may include:
- Failure to establish and implement written procedures for quality control testing of finished products
- Lack of procedures for the control and handling of raw materials
- Inadequate procedures for the maintenance and calibration of equipment used in the manufacturing process
2. Poor record-keeping
Record-keeping is an essential part of ensuring compliance with FDA regulations. Poor record-keeping refers to a failure to maintain accurate, complete, and up-to-date records of manufacturing, testing, and distribution activities.
Examples of poor record-keeping may include:
- Failure to maintain records of equipment maintenance and calibration
- Incomplete or inaccurate records of manufacturing or testing activities
- Failure to retain records for the required time period
3. Lack of employee Training
Lack of employee training refers to a failure to provide employees with adequate training on FDA regulations, policies, and procedures. This can result in employees not understanding their roles and responsibilities, leading to non-compliance with regulations. Examples of lack of employee training may include:
- Failure to provide training on the handling and storage of raw materials and finished products
- Lack of training on quality control procedures and testing methods
- Insufficient training on safety procedures and the use of equipment
4. Contamination and cross-contamination
Contamination and cross-contamination refer to the presence of harmful substances or microorganisms in products or manufacturing environments. This can occur due to inadequate cleaning procedures, lack of hygiene, or improper handling of raw materials or finished products. Examples of contamination and cross-contamination may include:
- Presence of bacteria or other microorganisms in finished products
- Contamination of products with cleaning agents or other chemicals
- Cross-contamination of products due to improper handling of raw materials
5. Failure to follow written procedures
Following written procedures is critical to ensuring compliance with FDA regulations. Failure to follow written procedures can result in non-compliance and the production of products that do not meet FDA standards. Examples of failure to follow written procedures may include:
- Failure to follow written procedures for cleaning and sanitation of equipment and manufacturing areas
- Failure to follow written procedures for quality control testing of finished products
- Failure to follow written procedures for handling and storage of raw materials
2023 FDA warning letter & inspection trends
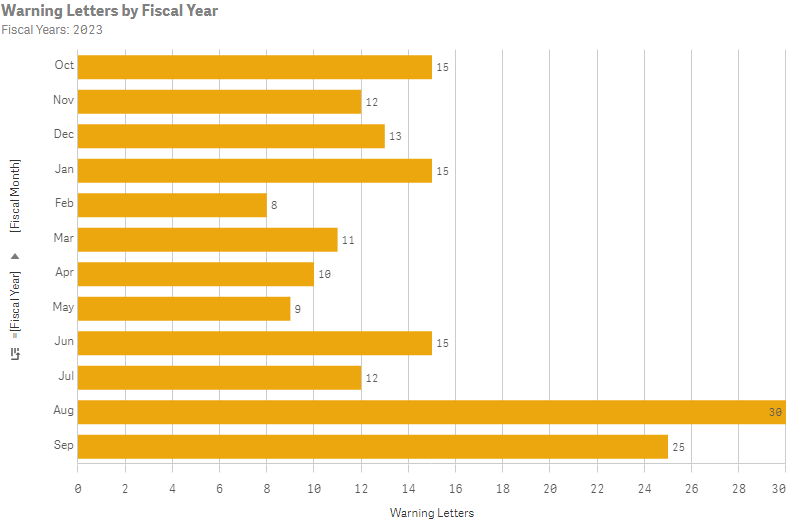
An examination of the citations in the inspection-related warning letters showed the FDA’s primary citations for the fiscal year 2023.
- FDA most often cited 21 CFR 211.100(a) for the lack of sufficient written procedures for production and process control, noting it 38 times in the 94 inspection-based warning letters examined. This citation is particularly prevalent among OTC manufacturers who have not performed process validation for their products.
- Equally cited as the most frequent, section 211.22, which addresses the failure of quality control to fulfill its duty in ensuring that drug products are manufactured in compliance with cGMP, was also mentioned 38 times. Specifically, subsections (a) and (d) of 21 CFR 211.22 were highlighted by the agency, underscoring the focus on shortcomings within quality units. This regulation has consistently been among the most commonly cited for several years, particularly concerning OTC firms, which frequently possess insufficient or no quality units at all.
- The FDA cited 21 CFR 211.84, which pertains to the failure to test and approve or reject components, drug product containers, and closures, a total of 37 times. Specifically, the agency frequently cited subsections (d)(1) (failure to verify the identity of each component of a drug product through testing) and (d)(2) (failure to appropriately validate and establish the reliability of a component supplier’s test analyses), often together. Although this regulation has not typically been among the top cGMP violations in the past, its prominence rose in FY23 due to the FDA’s increased attention to contaminants.
- 21 CFR 211.192, which deals with the failure to thoroughly investigate any unexplained discrepancy or a batch failure to meet specifications, whether distributed or not, was cited 30 times. Many of these violations involved the release of products despite out-of-specification (OOS) results. The agency’s recommended follow-up often included a critique that a retrospective review was not provided to ensure all OOS results and discrepancies were fully identified and thoroughly investigated. This regulation remains one of the most frequently cited, indicating ongoing challenges within the industry to conduct effective investigations.
- Concluding the top five most cited regulations is 21 CFR 211.165, noted for the failure to test products before their release for distribution, with 27 citations. Like 211.100, 211.22, and 211.192, this regulation consistently appears among the most frequently cited. It aligns with the FDA’s ongoing emphasis on OTC manufacturers and the presence of hazardous contaminants
Consequences of receiving form 483s and warnings
Not addressing a Form 483 or warning from the FDA can have serious consequences for a company. The potential consequences may include delays in the approval of new products, suspension or revocation of licenses, legal action, and damage to reputation.
- Delays in the approval of new products—The FDA may withhold approval of a new drug or medical device until the company can demonstrate that it has addressed the issues raised in the Form 483 or warning. This delay can result in lost revenue and market share.
- Suspension or revocation of licenses—If the FDA determines that a company has not taken adequate steps to address the issues raised in the Form 483 or warning, it may suspend or revoke the company’s licenses to manufacture or distribute products.
- Legal action—The FDA may take legal action against a company if it determines that the company has violated FDA regulations. This can result in fines, penalties, and other legal consequences.
- Damage to a company’s reputation—Negative publicity surrounding FDA inspections and violations can erode consumer confidence in a company’s products and lead to a decline in sales.
It is important for companies to understand the most common reasons for receiving Form 483s and warnings to ensure they are compliant with FDA regulations and avoid potential regulatory action. By doing so, companies can avoid the consequences associated with receiving Form 483s and warnings and ensure the safety and efficacy of their products.
Several companies have suffered consequences due to Form 483s and warnings. For example, in 2019, the FDA issued warning letters to five companies who produce products labeled as homeopathic for significant violations of current good manufacturing practice (CGMP) regulations, leading to product recall and damage to reputation.
In 2021 a medical device company received an FDA warning following a facility inspection. The company said the warning letter “focuses on the inadequacy of specific medical device quality system requirements at the Northridge facility in the areas of risk assessment, corrective and preventive action, complaint handling, device recalls, and reporting of adverse events.” The company’s stock price dropped by almost 9% following the announcement, as the company faced scrutiny from investors and regulatory agencies.
How to avoid receiving form 483s and warnings
Receiving a Form 483 or warning from the FDA can have serious consequences for a company. However, there are steps that companies can take to avoid receiving these citations.
One tip for avoiding Form 483s and warnings is to conduct regular self-audits. Companies should regularly review their operations and procedures to identify potential areas of non-compliance with FDA regulations. This can help companies address issues before they become serious problems and demonstrate to the FDA they take compliance seriously.
Ensuring procedures are up-to-date and followed is another key step in avoiding Form 483s and warnings. Companies should regularly review their procedures to ensure they comply with FDA regulations and reflect best practices in the industry. Employees should be trained in all relevant FDA regulations, as well as in the company’s procedures for complying with these regulations and that they are followed consistently. This training should be ongoing and should be updated as regulations and procedures change.
Maintaining accurate and complete records is also critical for avoiding Form 483s and warnings. Companies should have systems in place to ensure that records are accurate, complete, and up to date. This includes records related to manufacturing, quality control, validation, and distribution.
When dealing with data, you want to know when the record was created and who created it. If it was modified, who modified it, when they modified it, and why they modified it. Our digital validation solution Kneat Gx automatically generates a detailed, un-editable audit trail in real time. You can see any user action, correction or change with a corresponding timestamp, and much more at the click of a mouse and you can filter and re-order any audit trail to pinpoint any desired information during audit.
Companies should have systems in place to identify and address potential issues as soon as they arise. This can include conducting investigations into the root causes of issues and implementing corrective and preventive actions to prevent them from recurring.
Final thoughts
Compliance with FDA regulations is essential for companies operating in the pharmaceutical, biotechnology, and medical device industries. The consequences of receiving a Form 483 or warning can be severe, and companies should take proactive steps to avoid these violations. By prioritizing compliance and taking steps to ensure that their operations are in line with FDA regulations, companies can protect their reputation and avoid the negative consequences of non-compliance.
Learn more
When digitizing validation records, it is important to look beyond simply achieving compliance. You should select and apply the right software that helps you build a modern, data-centric approach to your validation program while offering broader value creation, including audit readiness.
Book a live demonstration of Kneat Gx, highlighting its key features, benefits, and how it can be used to streamline your validation processes, increase efficiency, and support regulatory compliance in your organization—just like it does for eight of the world’s top 10 pharmaceutical companies today.



