
What to leverage and what to own
When regulated companies approach computerized system validation, one of the most common — and most costly — mistakes is treating vendor documentation as something to validate around, rather than something to validate with. The result is duplicated effort, inflated timelines, and a validation program that proves little while consuming a lot.
FDA Computer Software Assurance (CSA) guidance and the second edition of ISPE GAMP 5 both point in the same direction: leverage what suppliers have already done, scale your effort to the risk, and apply critical thinking at every stage. But leveraging supplier activities doesn’t mean rubber-stamping vendor documentation. It means making informed, defensible decisions and documenting where your regulated responsibilities begin and seeing what objective evidence from the supplier can be leveraged.
This article walks through the key principles and practical considerations for getting that balance right.
Why critical thinking is the foundation

Before any conversation about what to leverage, it’s worth establishing why critical thinking is the starting point — not a nice-to-have, but a requirement embedded in the guidance itself.
GAMP 5 Second Edition defines critical thinking as promoting “informed decision-making and good judgement on where and how to apply and scale quality and compliance activities for computerized systems.” It also states that critical thinking “should be applied to understand the implementation risks as well as functional risks to patient safety, product quality, and data integrity.”
Critically, GAMP 5 is explicit that regulated companies must apply critical thinking during supplier assessment. That means you need to understand your supplier’s approach to IT and information systems — not just accept their output.
The ICH Q9 framework reinforces this: risk evaluation should be grounded in scientific knowledge and ultimately tied to patient protection. The level of effort, formality, and documentation in your quality risk management process should be commensurate with the level of risk involved.
In practice, this means no two systems will require the same validation approach, and no amount of guidance or templates can substitute for the informed judgement of a qualified team.
Determining who does what
Before testing begins, every team needs clarity on five questions:
- What deliverables do we need?
- What are we responsible for producing?
- What is the vendor responsible for?
- Who manages changes, and how?
- Where are all of the above formally documented?
These questions aren’t rhetorical. The answers should be captured in a Validation Plan that explicitly documents intent to leverage supplier activities, and in a Quality Agreement that binds both parties to defined responsibilities.
Without this upfront alignment, validation programs tend to drift — either duplicating work the vendor has already completed, or leaving gaps that auditors will find.
Assessing and selecting a supplier
Supplier assessment is a risk-based activity. The depth of your evaluation should reflect the risk that the system poses to patient safety, product quality, and data integrity.
GAMP 5 identifies several dimensions of supplier assessment: commercial reputation, quality systems, development and testing practices, and ongoing audit schedules. Regulated companies should build supplier audits into their supplier audit schedule and revisit them regularly — not just at the point of onboarding.
One practical point worth emphasizing: a supplier audit is not a one-time checkbox. It is a recurring quality activity that informs how much you can rely on supplier documentation across the system lifecycle.
What leveraging supplier activities actually means
Both the FDA CSA Guidance and GAMP 5 Second Edition give clear direction on leveraging supplier work — and both are careful about what that means in practice.
The FDA CSA guidance notes that for supporting software that carries lower risk, assurance activities may be reduced. Where supporting software’s performance is inherently covered by direct-use assurance activities, additional scripted or unscripted testing may be unnecessary. Manufacturers remain responsible for determining what assurance activities are needed to maintain the validated state.
GAMP 5 goes further, describing a principle of maximizing supplier involvement throughout the system lifecycle: “Regulated companies should seek to maximize supplier throughout the system lifecycle in order to leverage knowledge, experience, and documentation, subject to satisfactory supplier assessment.”
The key phrase is “subject to satisfactory supplier assessment.” Leverage is earned through a documented evaluation of the supplier’s quality system — not assumed because the vendor is well-known or commercially successful.
GAMP 5 also addresses documentation directly: supplier documentation should be assessed for suitability and completeness, with flexibility regarding format and structure. Regulators, the guidance notes, look for “scaled paperwork with well-organized information and records that have an appropriate level of detail, supported by clear and unambiguous rationales explaining critical thinking applied.”
When vendor documentation isn’t enough
A critical nuance — one that often gets lost in the enthusiasm for reducing validation effort — is that leveraging vendor documentation is not the same as accepting it unconditionally.
GAMP 5 is direct: “It’s not always the case that you just accept that what the Vendor has is enough.” If a vendor’s processes do not satisfy the deliverables of your Quality System as defined by your validation approach, you may need to perform certain tasks yourself. As the end user, you carry the regulatory responsibility.
This is where critical thinking becomes most important. If a supplier’s test documentation doesn’t map to your system’s intended use, or if their risk assessment doesn’t address your specific configuration, you need to supplement — not just accept what’s available.
Validation plans and vendor documentation
A well-constructed Validation Plan should document intent to leverage supplier activities from the outset. A Validation Plan should address at minimum:
- Functional specifications
- Product risk assessments
- Installation and functional testing records
- Traceability matrices and reports
- Testing responsibilities — what you will test versus what the vendor has tested or will test
- The sequence of testing activities, including Factory Acceptance Testing (FAT) and Site Acceptance Testing (SAT)
Defining these up front prevents the common situation where regulated companies discover late in a project that vendor documentation doesn’t match their internal requirements — and are left scrambling to fill gaps.
Quality plans and quality agreements

Quality plans and quality agreements serve related but distinct purposes.
A Quality Plan describes how the project management team will implement quality policy for a specific project. It should address quality control, quality assurance, and quality improvement activities, and should include provisions for supplier audits. Quality plans should be independently peer-reviewed and approved. Their formality and detail can vary — but the key is that they reflect the actual risk profile and responsibilities of the project.
The components of a strong Quality Management Plan include:
- Applicable QA and industry standards
- Quality objectives for the project
- Defined quality roles and responsibilities
- Clear acceptance criteria
- A list of deliverables and processes subject to quality review
- Quality control and management activities planned
- Quality methods to be applied
- Major procedures for handling non-conformance, corrective actions, and continuous improvement
A Quality Agreement, by contrast, is a binding document that defines commitments and responsibilities between a regulated company and a vendor for GxP use. It governs the entire relationship — not just the initial validation phase.
Support, maintenance, and change management
Validation doesn’t end at go-live. Support and maintenance introduce ongoing risk that needs structured evaluation.
GAMP 5 states that the supplier should support and maintain the system in accordance with agreed contracts. When changes or updates are deployed by the vendor, a risk-based approach should be taken to determine the appropriate response. Key questions include:
- Has the system’s intended use changed substantially?
- Have the changes increased or decreased risk associated with a particular feature or operation?
- Has new functionality been introduced, and has the supplier tested it to the appropriate level?
These questions should be asked systematically — not case by case based on whoever happens to notice a release note.
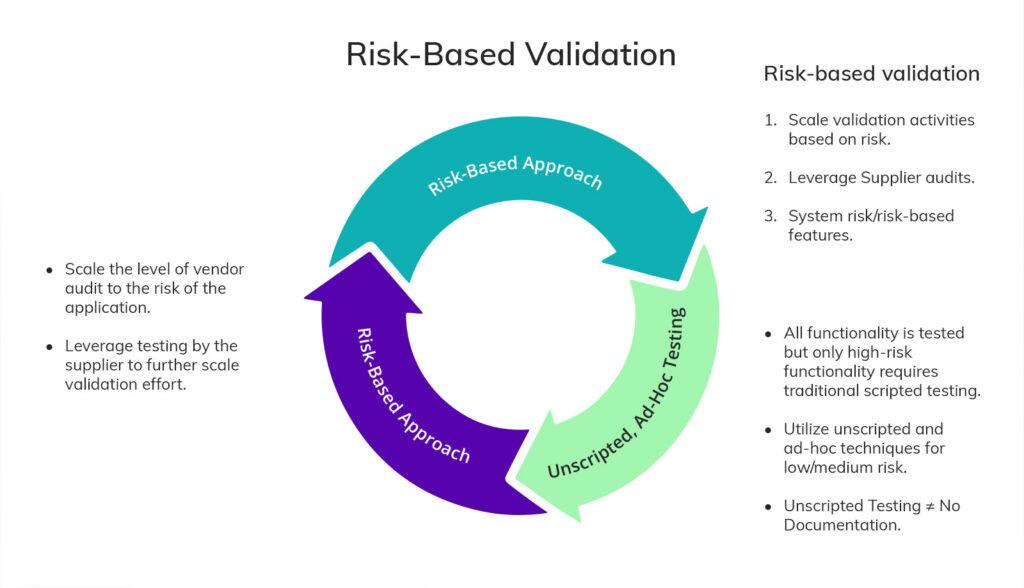
Risk-based validation in practice
Bringing these principles together, a risk-based validation approach for computerized systems works as follows:
All functionality is tested — but only high-risk functionality requires traditional scripted testing. Lower- and medium-risk functionality can use unscripted and ad-hoc techniques. Unscripted testing is not the same as no documentation. It means proportionate documentation, not absent documentation.
Vendor audits are scaled to risk. The depth of your audit should reflect the application’s risk profile. Supplier testing can be leveraged to further reduce your own validation effort — but only where the supplier’s quality system has been assessed as suitable.
Customer-specific requirements are assessed separately. Vendor documentation covers the product as designed. Your specific configuration, business process, and regulatory environment introduce additional requirements that must be evaluated and addressed independently.
Main takeaways
- Apply critical thinking throughout — from supplier selection through ongoing support. The guidance is explicit that this is a required competency, not an optional one.
- Leverage existing supplier activities and data. Do not duplicate work that has already been done to an adequate standard. Take credit for supplier effort where it is justified.
- Don’t assume vendor documentation is automatically sufficient. Assess it against your Quality System requirements and supplement where needed. You remain the responsible party from a regulatory standpoint.
- Involve subject matter experts (SMEs) when applying critical thinking to supplier assessment and validation planning. This is not work that should default to process alone.
See how Kneat Gx supports your CSV and CSA approach
Kneat Gx is built for the way regulated companies validate today — risk-based, scalable, and designed to leverage supplier documentation without the duplication. See it in action with a personalized demo.



