
If you work in quality, validation, IT, or engineering within the life sciences industry, chances are you’ve heard of Computer System Validation – probably by its initialism, CSV. Computer System Validation is the process of ensuring software and computer systems do what they’re intended to when they impact human health or product quality in life sciences manufacturing. CSV is a regulated requirement mandated by the U.S. FDA, under 21 CFR 11 and in Europe, under EudraLex Annex 11.
Is that all you need to know? Absolutely not, but it’s a start. In this article we’ll explore CSV in depth, examine its challenges, explain why the FDA is asking companies to do less work on it, and look at how you can do CSV and validation better.
The FDA and computer system validation
The U.S. Food and Drug Administration (FDA) regulates several industries, notably the pharmaceutical, medical device, and biotechnology industries, together considered Life Sciences. The FDA’s scope is broad and includes the tools and systems life sciences manufacturers may use in production, including computer software. The FDA mandates, under 21 CFR Part 11 Section 11.10, that software and computer systems are tested for consistency and efficacy and that those tests are documented and provable.
FDA penalties for non-compliance in Computer System Validation may include audits, warning letters, or even shutdowns if linked to significant risk in product quality.
FDA requirements for CSV validation
The FDA regulations mandate that CSV processes:
- Ensure the accuracy, reliability, and consistency of performance for software. As well as the ability to detect invalid or altered records.
- Can generate accurate and complete copies of records for suitable inspection, review, and copying by the FDA.
- Protect and store records.
- Use secure, computer-generated, time-stamped audit trails when records are created, modified, or deleted. These audit trails are to be maintained.
- Have operational system checks to enforce correct sequencing of steps as required. (Essentially the process itself is validated to ensure it is effective and followed.)
- Enforce access to computer systems and records so only authorized personnel can use, sign, or operate the system and records.
- Check to ensure input data or operational instructions are valid.
- Determine that personnel who develop and execute the record keeping, testing, and approvals have the proper experience and training.
- Use appropriate revision and change control procedures to maintain audit trails.
EMA, EudraLex Annex 11 and computer system validation
Where the FDA regulates life sciences (among others) manufacturing for U.S. consumers, the EU has different means of ensuring product safety and quality. The equivalent regulation to the FDA 21 CFR Part 11 for the EU is EudraLex Volume 4, which concerns Good Manufacturing Practice in Medicinal Products for Human and Veterinary Use.
Annex 11: Computerised Systems of EudraLex Volume 4 applies to “all forms of computerised systems used as part of a GMP regulated activities.” It mandates the validation and testing to ensure such systems do not decrease the quality of products or increase overall risk of processes.
The regulation largely matches that outlined by the FDA. Validation activities under one typically meet the requirements of the other, however, a company doing business in a market must always be prepared to demonstrate compliance with that market’s regulations and be able to present to that market’s relevant authority.
Annex 11’s computer system validation expectations are:
- Validation documentation and reports should cover all relevant steps of the lifecycle. Manufacturers should be able to justify their standards, protocols, and acceptance criteria based on risk assessments.
- Validation documentation should include change control records and document deviations observed.
- Processes should include a timely list of all relevant systems and their functionality. Critical systems should also detail physical and logical arrangements, data flows, and interfaces with other systems, software, or hardware and be readily available.
- User Requirements Specifications (URS) should describe the necessary functions of the system and contain documented risk assessments and GMP impacts. URS documents should be traceable throughout a product’s lifecycle.
- Any company in scope should take all reasonable steps to ensure a system used in life sciences production is developed according to appropriate quality standards, i.e., vendors are vetted and assessed.
- Purpose-built systems should have processes to ensure formal assessment and reporting of quality and performance measures are in place throughout the entire lifecycle.
- Testing should be demonstrated through evidence and documented assessments.
- If data is transferred to another format or system, validation should include ensuring the data is not altered in value or meaning.
Challenges of computer system validation
As you can imagine, computer system validation requires significant testing and documentation. Paper-based validation has struggled to keep up with demands as more and more software systems enter the life sciences manufacturing sector.
Some challenges often associated with traditional computer system validation include:
- Large scope of testing.
- One-size-fits-all approach to various systems and tools.
- Heavy documentation requirements which required significant resources to execute and manage.
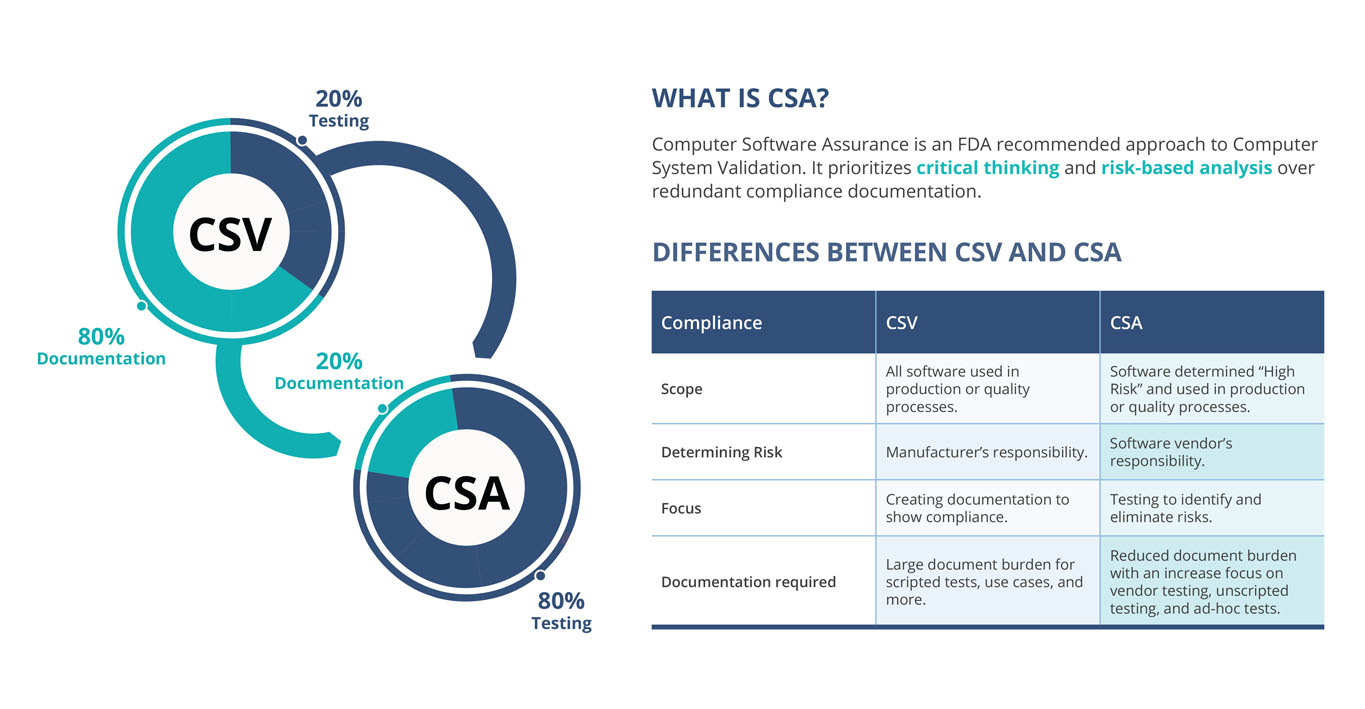
Computer software assurance (CSA)
In the 2011 Case for Quality, the FDA determined that the life sciences industry was slower to adopt new technologies than other sectors, believed in part due to the burdensome approach of CSV. In late 2022, the FDA issued draft guidance introducing Computer Software Assurance as the recommended approach to CSV.
At a high level, CSA is a risk-based approach that seeks to empower companies to perform CSV with activities that meet the risk of the systems in question, rather than implementing a one-size-fits-all approach. This includes leveraging vendor evidence rather than in-house work, and performing unscripted or ad hoc tests alongside more involved scripted tests to validate.
CSV testing
Traditional Computer System Validation used scripted testing for every in-scope system or tool. Scripted testing involved recording “actual” results, meaning complete details rather than a “pass/fail” recording. These would be documented alongside detailed test steps and screenshots where appropriate.
Under the FDA’s CSA guidance, companies can use unscripted and ad hoc testing to streamline CSV. We explore CSV testing in more detail here.
Digital validation to support CSV
When it comes to CSV and validation, the best way to execute is on a digital platform that eliminates paper, ensures compliance, and streamlines the entire validation lifecycle. Kneat’s CSV application has been proven to reduce validation time by over 50%.
Kneat Gx eliminates paper, enables online test execution of all types, seamless addition of screenshots and vendor tests, and is built to enable CSA’s risk-based approach to CSV validation. See it for yourself with a live demonstration during our upcoming webinar.



