
FDA 21 CFR Part 11 defines the criteria under which the FDA considers electronic records and electronic signatures to be trustworthy, reliable, and equivalent to paper records and handwritten signatures. The following key points summarize its scope, requirements, and relevance to life sciences organizations:
- Established in 1997, FDA 21 CFR Part 11 sets the criteria for electronic records and electronic signatures to be equivalent to paper records and handwritten signatures.
- Applies narrowly to records required under predicate rules that are maintained electronically in place of paper format.
- Enforced through specific controls including system access limits, operational checks, authority checks, device checks, user training, accountability policies, documentation controls, and electronic signature requirements.
- Fundamental to digital transformation in the pharmaceutical, biotechnology, and medical device sectors, where compliance ensures data integrity and regulatory confidence.
- Subject to FDA enforcement discretion on validation, audit trails, record retention, and record copying requirements, though predicate rules must still be met.
- Kneat Gx enables compliance by providing automated workflows, comprehensive audit trails, and secure electronic signatures — helping life sciences companies meet Part 11 requirements efficiently while driving productivity and innovation.
This guide outlines the key requirements of FDA 21 CFR Part 11 and explains how Kneat’s digital validation platform, Kneat Gx, supports life sciences organizations in achieving and maintaining compliance.
What is FDA 21 CFR Part 11?
FDA 21 CFR Part 11 sets forth criteria under which the FDA considers electronic records, electronic signatures, and handwritten signatures executed to electronic records to be trustworthy, reliable, and equivalent to paper records, enacted in 1997. The regulation was intended to permit the widest possible use of electronic technology, compatible with FDA’s responsibility to protect public health.
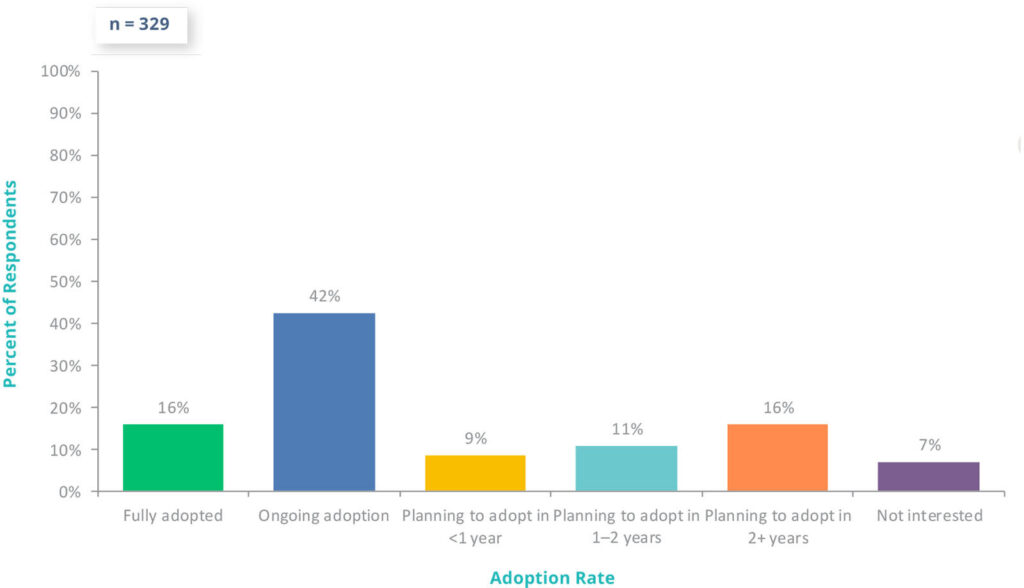
For pharmaceutical, biotech, and medical device companies, Part 11 compliance is fundamental to digital transformation. With FDA’s increasing scrutiny of data integrity, 58% of companies are already using digital validation systems in 2025, and an additional 35% plan to adopt one in the next two years.
Kneat’s perspective
Kneat Gx helps organizations achieve compliance with Part 11 while unlocking additional benefits for validation professionals, such as enhanced data integrity and smarter documentation collaboration and control. Kneat Gx scales with company growth and new regulatory guidance such as Computer Software Assurance (CSA), enabling global team collaboration in real time with automated notifications and in-line commenting capabilities.
Quick definition
Part 11 provides criteria for acceptance by FDA of electronic records, electronic signatures, and handwritten signatures executed to electronic records.
Scope and purpose
Industries Covered: FDA regulates pharmaceutical, medical device, and biotechnology industries, with Part 11 applying to the tools and systems manufacturers use in production, including computer software.
Geographic Reach: While FDA regulates for US consumers, the EU guideline aligned with 21 CFR Part 11 is EudraLex Volume 4 Annex 11, and validation activities under one typically meet requirements of the other.
Lifecycle Phase: Part 11 applies to records in electronic form that are created, modified, maintained, archived, retrieved, or transmitted under any records requirements set forth in Agency regulations.
Regulatory authority / Issuing body
The guidance was prepared by the Office of Compliance in the Center for Drug Evaluation and Research (CDER) in consultation with other FDA centers and the Office of Regulatory Affairs.
History and key revisions
| Date | Event |
|---|---|
| 1991 | FDA establishes task force for electronic records approach |
| March 1997 | FDA issues final Part 11 regulations |
| August 1997 | Part 11 becomes effective |
| 2003 | FDA announces enforcement discretion guidance for validation, audit trails, record retention, and record copying |
Some confusion persists today regarding the 2003 guidance, as the FDA stated it would “execute enforcement discretion with respect to certain Part 11 requirements” without further clarification.
Kneat’s perspective
Despite regulatory evolution, the purpose of Part 11 remains hugely relevant and has grown in importance over 25+ years as more life sciences companies employ new technological solutions to streamline validation work.
Kneat’s document control features meet FDA requirements for electronic records and signatures while streamlining validation workflows, empowering regulators with oversight confidence and enabling companies to leverage technology safely for competitive growth.
Key requirements of FDA 21 CFR Part 11
Electronic records requirements
| Requirement | Description |
|---|---|
| System Access Controls | Limiting system access to authorized individuals |
| Operational System Checks | Use of operational system checks |
| Authority Checks | Use of authority checks |
| Device Checks | Use of device checks |
Electronic signatures requirements
Electronic signatures must be equivalent to handwritten signatures, initials, and other general signings required by predicate rules, used to document that certain events or actions occurred in accordance with predicate rules (e.g., approved, reviewed, verified).
Organizations must establish and adhere to written policies that hold individuals accountable for actions initiated under their electronic signatures.
Documentation and system controls
- Appropriate controls over systems documentation
- Controls for open systems corresponding to controls for closed systems
- Requirements related to electronic signatures per §§ 11.50, 11.70, 11.100, 11.200, and 11.300
Validation
The FDA exercises enforcement discretion regarding specific Part 11 validation requirements (§ 11.10(a)), though persons must still comply with all applicable predicate rule requirements for validation.
Validation decisions should be based on justified and documented risk assessment considering impact on predicate rule requirements.
Audit trails
The FDA exercises enforcement discretion on computer-generated, time-stamped audit trails (§ 11.10(e), (k)(2)), but persons must comply with predicate rule requirements for documentation of date, time, or sequencing of events.
Even without predicate rule requirements, audit trails or other security measures may be important to ensure record trustworthiness, particularly when users create, modify, or delete regulated records during normal operation.
Record retention
The FDA exercises enforcement discretion on Part 11 requirements for record protection and retrieval (§ 11.10(c)), though persons must comply with all applicable predicate rule requirements for record retention and availability.
FDA does not object to archiving required records to nonelectronic media (microfilm, microfiche, paper) or standard electronic formats (PDF, XML, SGML), provided predicate rule requirements are satisfied and content and meaning are preserved.
Why compliance matters
Regulatory penalties and enforcement
Failure to comply can lead to product recalls, significant reputational damage, and even criminal penalties.
FDA penalties for non-compliance in Computer System Validation may include audits, warning letters, or even shutdowns if linked to significant risk in product quality.
FDA enforcement actions can result in facility shutdowns, product recalls, delayed or denied approvals, import and distribution bans, criminal prosecution, remediation costs, and loss of customers due to damaged reputation.
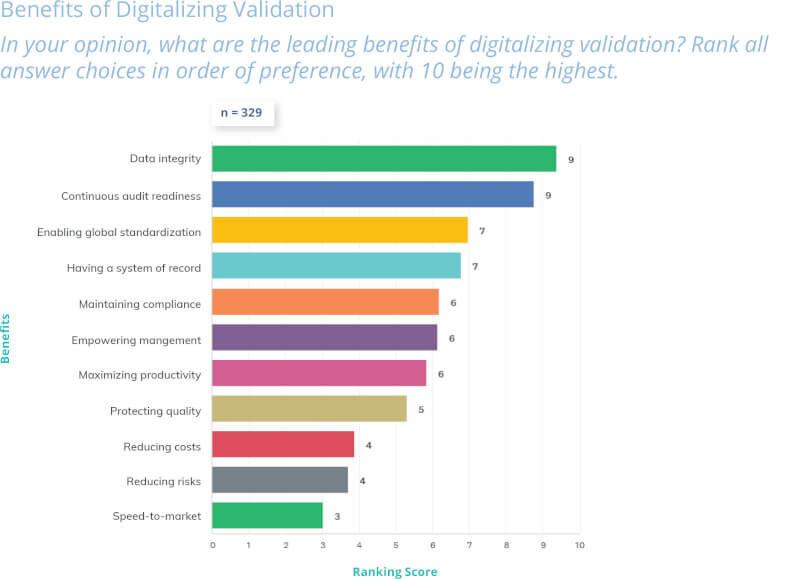
Of the 75 warning letters issued by the FDA in 2016, 43% were linked to data integrity violations, rising to 60% in 2017.
Business and patient impact
Part 11’s contribution to compliance benefits all stakeholders: regulators gain oversight confidence, companies leverage technology safely for competitive growth, and required systems result in better process control, information transfer, data integrity, and fewer data-related errors.
Patients benefit from faster innovations enabled by digitalization and faster data analysis, while consumer confidence is promoted through preservation of data integrity.
Kneat’s perspective
Kneat’s digital validation solution builds data integrity into every step of the validation lifecycle with automatic time stamps and audit trails, password-protected electronic signatures, and paperless workflows.
Kneat captures all validation data directly to a secure database with comprehensive time-stamped audit trails, enabling Part 11 Subpart C (§§11.50–11.300) compliant signatures and secure role-based user access control.
Step-by-step compliance roadmap
Gap assessment
Determine based on predicate rules whether specific records are Part 11 records and document such decisions.
For each record required under predicate rules, determine in advance whether you plan to rely on the electronic or paper record to perform regulated activities and document this decision in SOPs or specification documents.
Process and technology controls
Part 11 applies narrowly: only when persons choose to use records in electronic format in place of paper format for records required under predicate rules.
Implement the enforced controls:
- System access limits, operational checks, authority checks, device checks
- User education, training, and experience requirements
- Written accountability policies for electronic signatures
- Documentation controls and open/closed system controls
Documentation best practices
Thorough documentation of all aspects of system validation, including testing protocols, results, and deviations, provides evidence of compliance and facilitates regulatory inspections.
Supply copies of electronic records by producing copies in common portable formats or using established automated conversion methods to formats like PDF, XML, or SGML.
Ongoing monitoring and audit prep
Compliance with Part 11 is an ongoing process requiring regular monitoring and maintenance activities to ensure systems remain in a validated state and continue to meet regulatory requirements.
Strong data and document management processes are essential to audit readiness, whether for audits, inspections, or remote regulatory assessments.
Kneat’s perspective
Good validation software ensures audit-readiness by conducting every part of the process within the system, with validation activities linked in real time to requirements, risks, and protocols, with every step digitally logged.
Kneat’s centrally stored data and documents can be automatically tracked using Real-Time Requirements Traceability Matrix (RTM), which supports audit-readiness, making it easy to trace, find, and access documents as soon as needed during audits or inspections.
Common pitfalls and how to avoid them

1. Overly broad interpretation of scope
Pitfall: Some interpret Part 11 scope too broadly, leading to unnecessary controls and costs that discourage innovation without added public health benefit.
Pro Tip: When persons use computers to generate paper printouts that meet all predicate rule requirements and rely on paper records for regulated activities, the FDA does not consider this “using electronic records in lieu of paper records.” Kneat helps clarify which records require Part 11 controls through risk-based assessment tools.
2. Inadequate system validation
Pitfall: Organizations may under-validate systems or impose unnecessary validation requirements.
Pro Tip: Base validation decisions on a justified and documented risk assessment considering impact on predicate rule requirements and on accuracy, reliability, integrity, availability, and authenticity of required records. Kneat’s platform is validated using Kneat-provided IQ/OQ documentation, requiring customer PQ validation for intended use.
3. Missing or insufficient audit trails
Pitfall: Organizations may fail to implement adequate audit trails despite predicate rule requirements.
Pro Tip: Even without explicit predicate rule requirements, audit trails ensure record trustworthiness and are particularly appropriate when users create, modify, or delete regulated records. Kneat provides comprehensive, time-stamped audit trails that capture all entries and changes.
4. Inadequate electronic signature controls
Pitfall: Electronic signature systems lack unique identification or accountability mechanisms.
Pro Tip: Part 11 compliant systems must have password and security features that limit user access and privileges, with unique electronic signatures assigned to each user. Kneat ensures users apply Part 11 Subpart C (§§11.50–11.300) compliant signatures with robust attribution and secure role-based access control.
5. Poor record retention strategy
Pitfall: Organizations fail to enable accurate and ready retrieval throughout required retention periods.
Pro Tip: Base record retention decisions on predicate rule requirements, justified risk assessment, and determination of record value over time. Kneat’s cloud-based system ensures secure, accessible storage with configurable retention policies aligned to regulatory requirements.
6. Legacy system non-compliance
Pitfall: Organizations continue using systems operational before August 20, 1997, without documenting fitness for intended use.
Pro Tip: The FDA exercises enforcement discretion for legacy systems if: (1) operational before effective date, (2) met all predicate rules before effective date, (3) currently meets all predicate rules, and (4) you have documented evidence the system is fit for intended use. Migrating to Kneat provides a compliant, modern alternative that eliminates legacy system risks.
7. Inadequate record copies for inspections
Pitfall: Organizations fail to provide reasonable and useful access to records during inspections.
Pro Tip: Supply copies in common portable formats (PDF, XML, SGML) that preserve content and meaning, with same search, sort, and trend capabilities if reasonable and technically feasible. Kneat’s RTM makes it easy to provide inspectors with requested documents instantly.
FAQs
What is the main goal of FDA 21 CFR Part 11?
The main goal is to establish criteria under which FDA considers electronic records, electronic signatures, and handwritten signatures executed to electronic records to be trustworthy, reliable, and equivalent to paper records. The regulations were intended to permit the widest possible use of electronic technology, compatible with FDA’s responsibility to protect public health.
Does FDA 21 CFR Part 11 apply to SaaS/Cloud systems?
Yes. Part 11 applies to records in electronic form that are created, modified, maintained, archived, retrieved, or transmitted under any records requirements set forth in Agency regulations. When persons choose to use records in electronic format in place of paper format for records required under predicate rules, Part 11 applies. Kneat’s cloud-based software scales with company growth and new regulatory guidance such as Computer Software Assurance (CSA), enabling global team collaboration.
How often is re-validation required under FDA 21 CFR Part 11?
The FDA exercises enforcement discretion on specific Part 11 validation requirements, though persons must comply with all applicable predicate rule requirements for validation. Compliance requires regular monitoring and maintenance activities to ensure systems remain in a validated state. Re-validation frequency depends on system changes, risk assessment, and predicate rule requirements rather than a fixed schedule.
Can electronic signatures satisfy FDA 21 CFR Part 11?
Yes. Electronic signatures that are intended to be equivalent to handwritten signatures, initials, and other general signings required by predicate rules satisfy Part 11, when used to document that certain events or actions occurred in accordance with predicate rules. Part 11 compliant systems must assign unique electronic signatures to each user that are legally equivalent to handwritten signatures.
Recent updates and future outlook (2024-2025)
While the core Part 11 regulation from 1997 remains in effect, the regulatory landscape continues to evolve:
Computer Software Assurance (CSA): In September 2025, the FDA issued its final Computer Software Assurance for Production and Quality System Software guidance, replacing the 2022 draft. The final guidance establishes a risk-based approach for establishing and maintaining confidence that software is fit for its intended use throughout its lifecycle. By focusing validation efforts on areas that pose the greatest risk to product quality and patient safety, the CSA framework supports a least burdensome, efficient approach to computer system validation that fosters innovation and the adoption of advanced technologies across life sciences manufacturing and quality systems.
Digital Transformation Trends: FDA’s increasing scrutiny of data integrity across the industry has led 93% of highly regulated companies to plan adoption of or currently use digital validation solutions.
Enforcement Focus: Greater scrutiny from global regulatory bodies has increased the likelihood of enforcement action related to electronic systems and data integrity.
Organizations should monitor FDA announcements through the Federal Register and industry guidance from organizations like the International Society for Pharmaceutical Engineering (ISPE) and Good Automated Manufacturing Practice (GAMP) for updates to Part 11 interpretation and enforcement. Kneat Gx scales with new regulatory guidance such as the FDA’s Computer Software Assurance (CSA), ensuring your validation platform evolves with regulatory requirements.
Looking Ahead: The FDA’s 2025 guidance on CSA aims to reduce the burden of CSV, improve quality, remove non-value added activities, and focus testing on high-risk areas only, eagerly awaited by validation professionals. This shift toward risk-based, efficient compliance aligns perfectly with Kneat’s platform capabilities, positioning our customers for success in the evolving regulatory landscape.
Need help achieving FDA 21 CFR Part 11 compliance? Partner with Kneat to discover how our digital validation platform Kneat Gx streamlines compliance while accelerating your validation lifecycle.



